



Cat I Don’t Like Moring People Or Mornings Or People Shirt
Or buy produyct at :Amazon
♥CHECK OUR BESTSELLERS - LIMITED EDITION SNEAKER FOR MEN OR WOMEN:


Best Selling Sneaker
Retro SP x J Balvin Medellín Sunset (UA) Air Jordan 3 Sneaker


Best Selling Sneaker
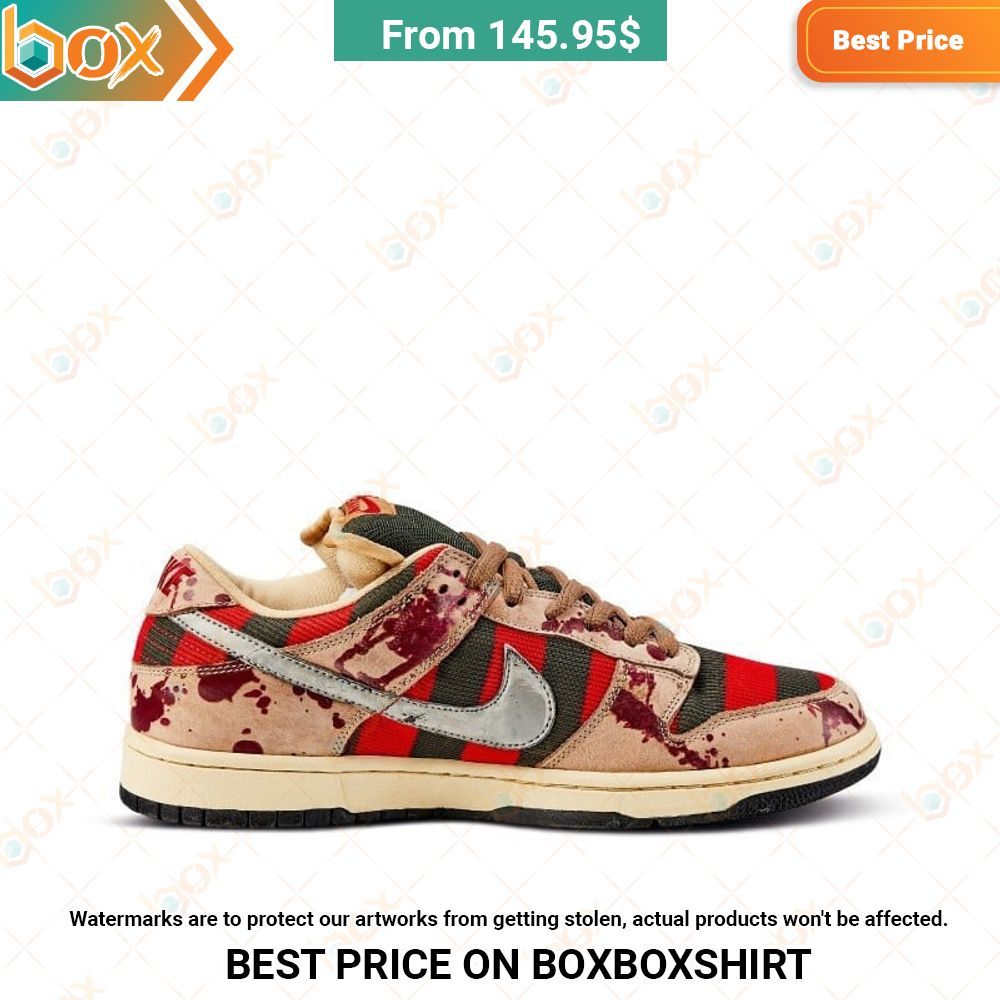

Best Selling Sneaker

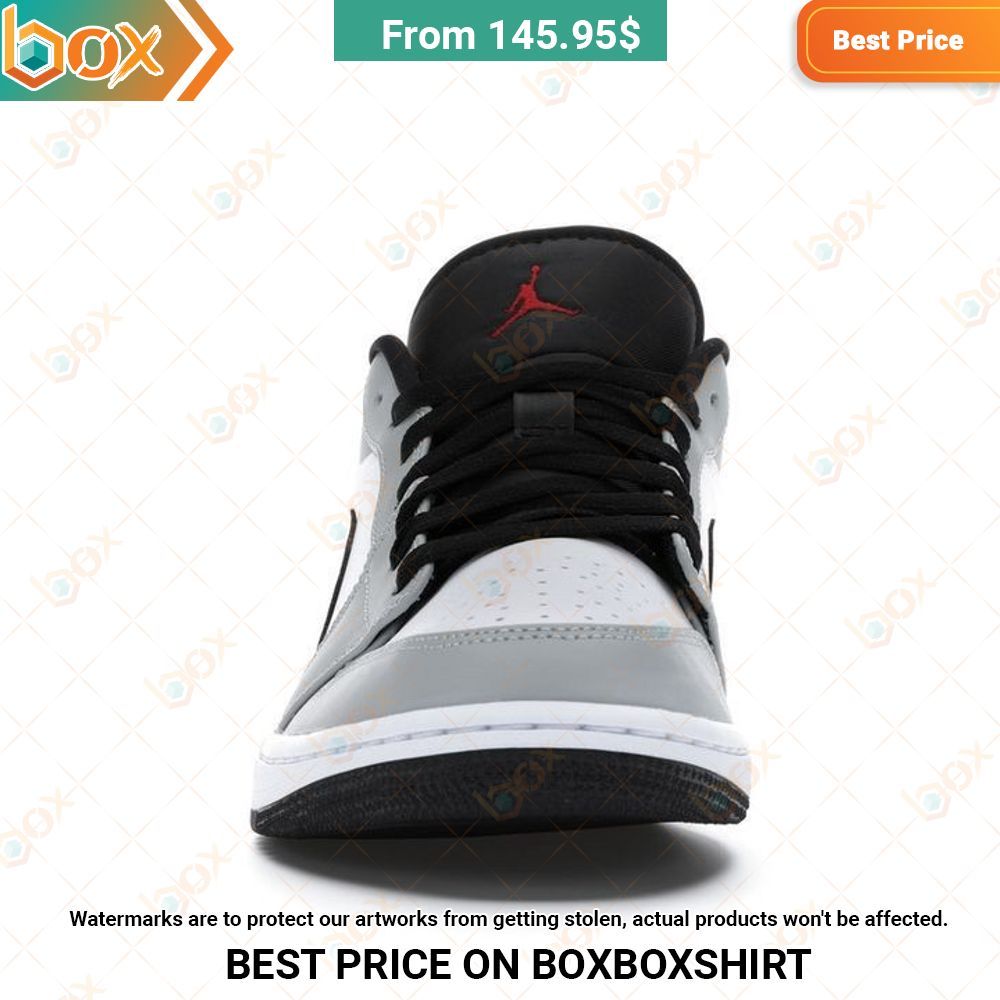
Best Selling Sneaker
Cat I Don’t Like Moring People Or Mornings Or People Shirt
Raw reads were quality-trimmed by Trimmomatic48 with a sliding window option, minimum base quality of 20 and minimum read length of 25 bp. The assembled polar bear genome23 was used for reference mapping using BWA version 0.7.5a49 with the BWA-MEM algorithm on scaffolds larger than 1 Mb. Scaffolds shorter than 1 Mb in length were not involved in the mapping and analyses, due to potential assembly artefacts50 and for reducing the computational time in downstream analyses. Duplicate Illumina reads were marked by Picard tools version 1.106 (http://picard.sourceforge.net/) and the genome coverage was estimated from Samtools version: 0.1.1851.Freebayes version 0.9.14–1752 called Single Nucleotide Variants (SNVs) using the option of reporting the monomorphic sites with additional parameters as -min-mapping-quality 20, -min-alternate-count 4, -min-alternate-fraction 0.3 and -min-coverage 4 with insertion/deletion (indel) realignment. A custom Perl script created consensus sequences for each of the mapped bear individuals from the Variant Call Format (VCF) files, keeping the heterozygous sites and removing indels. In order to complete the taxon sampling of the ursine bears, reads from six previously published genomes (Supplementary Table 1) selected and on the basis of geographic distribution, availability and sequence depth and SNVs were called as described above. For the two high coverage ( > 30X) genomes, SNVs calling parameters (-min-coverage) were set as one-half of the average read depth after marking duplicates. Genome error rates53 were calculated on the largest scaffold (67 Mb) for all bear genomes, confirming a high quality of the consensus sequences. (Supplementary Methods and Supplementary Fig. 22).
Cat I Don’t Like Moring People Or Mornings Or People Shirt
Data filtration, simulation of sequence length and topology testingThe next step was to create multi-species alignments for further phylogenetic analysis from all 13 bear individuals. In order to create a data set with reduced assembly and mapping artefacts, genome data was masked for TEs and simple repeats19 using the RepeatMasker54 output file of the polar bear reference genome available from http://gigadb.org/23. Since the polar bear reference genome RepeatMasker output file did not contain the simple repeat annotation, we repeatmasked the polar bear reference genome with the option (-int) to mask simple repeats. Next all bear genomes were masked with bedtools version 2.17.055 and custom Perl scripts. Non-overlapping, sliding window fragments of 100 kb were extracted using custom perl scripts together with the program splitter from the Emboss package56 (Supplementary Fig. 1), creating a dataset of 22,269 GFs from 13 bear individuals. Heterozygous sites, and repeat elements were all marked “N” and removed using custom Perl scripts. An evaluation of the minimum sequence length of GFs needed for phylogenetic analysis was done by estimating how much sequence data is needed to reject a phylogenetic tree topology using the approximate unbiased, AU test57. Only sufficiently long sequences can differentiate between alternative trees with statistical significance. The evaluation was done in two separate analyses: (a) with a simulated data set and (b) on a data set of 500 random GFs (Supplementary Methods).
A. SHIPPING COSTS
Standard Shipping from $4.95 / 1 item
Expedited Shipping from $10.95 / 1 item
B. TRANSIT, HANDLING & ORDER CUT-OFF TIME
Generally, shipments are in transit for 10 – 15 days (Monday to Friday). Order cut-off time will be 05:00 PM Eastern Standard Time (New York). Order handling time is 3-5 business days (Monday to Friday).
C. CHANGE OF ADDRESS
We cannot change the delivery address once it is in transit. If you need to change the place to deliver your order, please contact us within 24 hours of placing your order at contact.boxboxshirt@gmail.com
D. TRACKING
Once your order has been shipped, your order comes with a tracking number allowing you to track it until it is delivered to you. Please check your tracking code in your billing mail.
E. CANCELLATIONS
If you change your mind before you have received your order, we are able to accept cancellations at any time before the order has been dispatched. If an order has already been dispatched, please refer to our refund policy.
G. PARCELS DAMAGE IN TRANSIT
If you find a parcel is damaged in transit, if possible, please reject the parcel from the courier and get in touch with our customer service. If the parcel has been delivered without you being present, please contact customer service with the next steps.
No Hassle Returns and Refunds
Our policy lasts 14 days. If 14 days have gone by since your purchase, unfortunately we can’t offer you a refund or exchange.
To be eligible for a return, your item must be unused and in the same condition that you received it. It must also be in the original packaging.
Several types of goods are exempt from being returned.
Gift cards
Downloadable software products
Some health and personal care items
To complete your return, we require a receipt or proof of purchase.
Please do not send your purchase back to the manufacturer.
There are certain situations where only partial refunds are granted (if applicable) :
– Any item not in its original condition, is damaged or missing parts for reasons not due to our error
– Any item that is returned more than 30 days after delivery
Refunds (if applicable)
Once your return is received and inspected, we will send you an email to notify you that we have received your returned item. We will also notify you of the approval or rejection of your refund.
If you are approved, then your refund will be processed, and a credit will automatically be applied to your credit card or original method of payment, within a certain amount of days.
Late or missing refunds (if applicable)
If you haven’t received a refund yet, first check your bank account again.
Then contact your credit card company, it may take some time before your refund is officially posted.
Next contact your bank. There is often some processing time before a refund is posted.
If you’ve done all of this and you still have not received your refund yet, please contact us at contact.boxboxshirt@gmail.com








